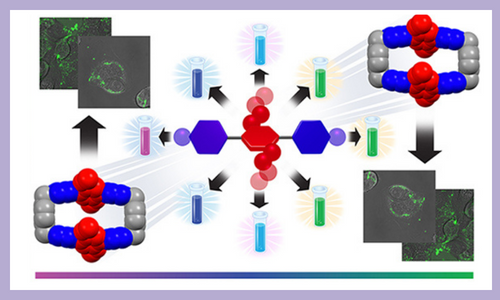
A rational approach to the design and synthesis of a series of fluorescent dyes that contain [acceptor-donor-acceptor] quadrupolar backbones is presented. The dyes have been functionalized with electron-rich donor rings in their midriffs and terminal acceptor rings of varying electron deficiencies, as dictated by the strength of the electron withdrawing groups. Their quadrupolarity-correlated photophysical properties includes a range of fluorescent colors which cover the visible spectrum from deep violet to green. Steady-state and transient absorption and emission spectroscopic analysis have been performed to gain deeper insight into intramolecular charge transfer. This investigation also demonstrates the non-cytotoxicity profile of one of the water-soluble dyes and obtain in vitro live-cell confocal microscopic images that show cell uptake at relatively low concentrations. J. Am. Chem. Soc. 144, 16841–16854 (2022)
|
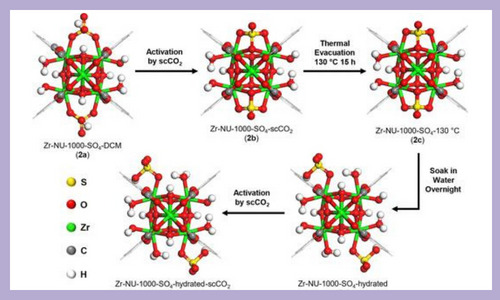
Understanding heterogeneous catalysts is a challenging pursuit due to surface site nonuniformity and aperiodicity in traditionally used materials. One example is sulfated metal oxides, which function as highly active catalysts and as supports for organometallic complexes. These applications are due to traits such as acidity, ability to act as a weakly coordinating ligand, and aptitude for promoting transformations via radical cation intermediates. Research is ongoing about the structural features of sulfated metal oxides that imbue the aforementioned properties, such as sulfate geometry and coordination. To better understand these materials, metal–organic frameworks (MOFs) have been targeted as structurally defined analogues. Composed of inorganic nodes and organic linkers, MOFs possess features such as high porosity and crystallinity, which make them attractive for mechanistic studies of heterogeneous catalysts. In this work, Zr6-based MOF NU-1000 is sulfated and characterized using techniques such as single crystal X-ray diffraction in addition to diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS). The dynamic nature of the sulfate binding motif is found to transition from monodentate, to bidentate, to tridentate depending on the degree of hydration, as supported by density functional theory (DFT) calculations. Heightened Brønsted acidity compared to the parent MOF was observed upon sulfation and probed through trimethylphosphine oxide physisorption, ammonia sorption, in situ ammonia DRIFTS, and DFT studies. With the support structure benchmarked, an organoiridium complex was chemisorbed onto the sulfated MOF node, and the efficacy of this supported catalyst was demonstrated for stoichiometric and catalytic activation of benzene-d6 and toluene with structure–activity relationships derived. J. Am. Chem. Soc. 144, 16883–16897 (2022)
|
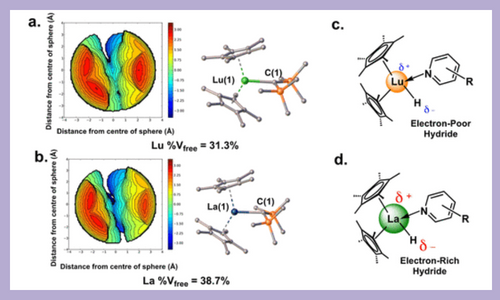
Chemodivergent synthetic methodologies enable efficient introduction of structural diversity of high-value organic products via simple chemical alterations. C–H activation and functionalization of pyridinoid azines are important transformations in creating many pharmaceuticals and natural products. Pyridinoid Azines contains nitrogen, which makes them difficult to activate. To facilitate activation, researchers discovered a method of using special metals called lanthanides to activate the molecule. Experimental and theoretical mechanistic data reported here support that changing the lanthanide identity and substrate substituent electronic character can make it easier to activate the molecule in various ways. This lanthanide series catalytic chemodivergence appears to be unprecendented. J. Am. Chem. Soc.144, 17086–17096 (2022)
|
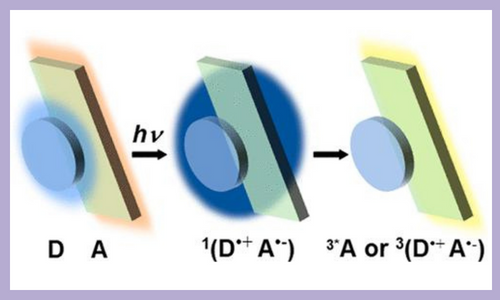
Ultrafast triplet formation in D-A, come together, triplet formation typically requires significant orbital angular momentum change thus it is usually observed when adjacent π systems of D and A are orthogonal. However, this study shows that subnanosecond triplet formation occurs in a series of D-A cocrystals that form something called a one-dimensional cofacial π stack. Utilizing ultrafast transient absorption microscopy to excite the molecules it was found that it took less than 300 femtoseconds for the charge transfer excitons to form. The triplet exciton formation took 125, 106, 484, and 958 picoseconds for each of the four different molecules studied. In addition, time-resolved EPR spectroscopy measured how much energy was released during the triplet formation. The results showed that the energy released correlated with how much charge was delocalized in the molecule. These results study show that that charge delocalization in CT excitons impacts both ultrafast triplet formation as well as the CT character of the resultant triplet states. J. Am. Chem. Soc. 144, 18607–18618 (2022) |
|
Two-dimensional (2D) covalent organic frameworks (COFs) are structurally precise, permanently porous, layered 2D polymers that are typically synthesized as aggregated, polycrystalline solids. This work represents the first example of polymerizing imine-linked 2D COFs as faceted single crystals in as little as five minutes at moderate temperature and ambient pressure. Single crystals of two imine-linked 2D COFs were prepared and characterized by electron diffraction as single-crystalline in the plane of covalent bonding with stacking disorder. Both single-crystalline COFs were incorporated into gas chromatography columns for molecular separations and significantly outperformed polycrystalline COFs, indicating the importance of improved materials quality for targeted applications. J. Am. Chem. Soc.144, 19813–19824 (2022)
|
|
Polymer networks built out of dynamic covalent bonds offer the potential to translate the control and tunability of chemical reactions to macroscopic physical properties. Under conditions at which these reactions occur, the topology of covalent adaptable networks (CANs) can rearrange, meaning that they can flow, self-heal, be remolded, and respond to stimuli. Materials with these properties are necessary to fields ranging from sustainability to tissue engineering; thus the conditions and time scale of network rearrangement must be compatible with the intended use. The mechanical properties of CANs are based on the thermodynamics and kinetics of their constituent bonds. Therefore, strategies are needed that connect the molecular and macroscopic worlds. In this Perspective, we analyze structure–reactivity–property relationships for several classes of CANs, illustrating both general design principles and the predictive potential of linear free energy relationships (LFERs) applied to CANs. We discuss opportunities in the field to develop quantitative structure–reactivity–property relationships and open challenges. J. Am. Chem. Soc. 144, 22358–22377 (2022)
|

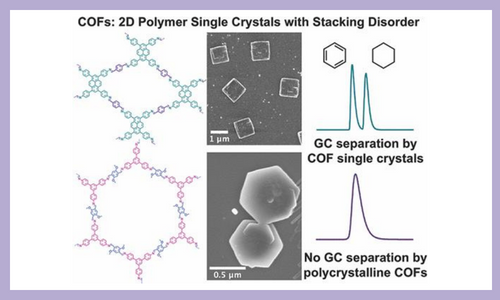 Single-Crystalline Imine-Linked Two-Dimensional Covalent Organic Frameworks Separate Benzene and Cyclohexane Efficiently
Single-Crystalline Imine-Linked Two-Dimensional Covalent Organic Frameworks Separate Benzene and Cyclohexane Efficiently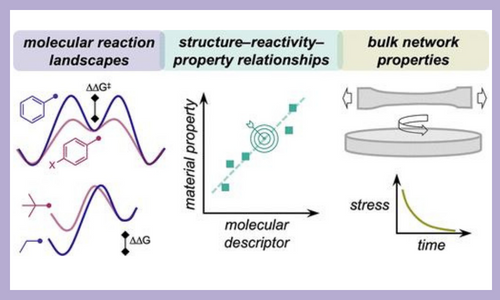 Structure–Reactivity–Property Relationships in
Structure–Reactivity–Property Relationships in